

Základní patologie
Fabryho choroba patří k více než padesáti známým vzácným dědičným onemocněním, označovaných jako lyzosomální poruchy. Každá z těchto poruch je způsobena vrozenou genetickou vadou, na jejímž základě dochází k nedostatku specifického lyzosomálního enzymu nebo enzymů. Nástup onemocnění, postižení orgánů a závažnost těchto poruch se výrazně liší, byť jsou všechny tyto poruchy progresivní.
Fabryho chorobu způsobuje mutace genu GLA, jenž kóduje lyzosomální alfa-galaktozidázu A (též známou jako alfa-GAL, alfa-Gal-A nebo ceramid trihexosidáza). Částečný nebo úplný deficit aktivity alfa-galaktozidázy A má za následek sníženou schopnost odbourávat lipidy obsahující zbytky konečného alfa-galaktozylu. Tyto lipidy, zejména globotriaosylceramid (známý také jako GL-3; Gb3, ceramid trihexosid nebo CTH), se hromadí v lyzosomech řady různých typů buněk v celém těle, včetně endoteliálních buněk kapilár, buněk v ledvinách, srdci a nervových buněk, s následným progresivním multisystémovým poškozením, což může časem vést až k úplnému poškození orgánů – ledvin, srdce a/nebo nervového systému.
Germain DP [online]. Orphanet J Rare Dis 2010 [cit. 08-08-2017]: http://dx.doi.org/10.1186/1750-1172-5-30
Hromadění GL-3 v endotelu kapilár
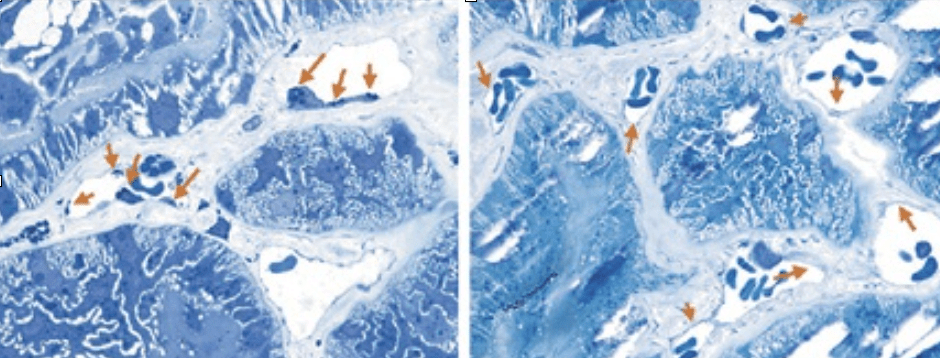
Šipky označují na elektronovém mikrografu místa v endotelu ledvinových kapilár, v nichž u pacienta s Fabryho chorobou dochází ke hromadění GL-3.
Obrázek byl použit se souhlasem Sanofi Genzyme.