

Progrese a prognóza
Proměnlivá hladina enzymu
Pompeho nemoc je charakterizována nižší aktivitou GAA, přesný stupeň reziduální enzymatické aktivity se však liší mezi jednotlivými pacienty i mezi populacemi pacientů:
- Kojenci s Pompeho nemocí zpravidla vykazují méně než 1 % průměrné normální aktivity GAA. van der Ploeg AT, Reuser AJ Lancet 2008;372(9646):1342-53.
- Děti a dospělí s Pompeho nemocí mohou vykazovat reziduální aktivitu GAA odpovídající 1−30 % průměrné normální hladiny. van der Ploeg AT, Reuser AJ Lancet 2008;372(9646):1342-53.
Obecně existuje jen slabá korelace mezi reziduální aktivitou GAA a klinickými projevy u dětí a dospělých, nicméně u kojenců je vždy přítomno těžké postižení.
Průběh onemocnění
Pacienti s klasickou infantilní formou Pompeho nemoci obvykle vykazují téměř úplnou absenci enzymatické aktivity GAA, mají výraznou kardiomegalii a rychlou akumulaci glykogenu v kosterním svalstvu, která může dosahovat až 10 násobku normálních hodnot. Hirschhorn R et al In: Scriver CR et al eds. The Metabolic & Molecular Bases of Inherited Disease 8th ed. New York, NY: McGraw-Hill; 2001:3389-3420. U této populace pacientů onemocnění postupuje velmi rychle a bez léčby je obvykle fatální v průběhu prvního roku života. Kishnani PS et al J Pediatr 2004;144(5 Suppl):S35-S43.
U dětí a dospělých nemoc obvykle postupuje pomaleji než u klasické infantilní formy a postižení srdce je malé nebo není přítomno. Nicméně Pompeho nemoc je progresivní a je spojena s významnou morbiditou a/nebo předčasnou mortalitou. Hagemans ML et al Brain 2005;128:671-7. Wokke J et al Muscle Nerve 2008;38:1236-45. Güngor D et al Orphanet J Rare Dis 2011; 6: 34.
Infantilní forma Pompeho nemoci je rychle progredující onemocnění, které má bez léčby zpravidla fatální následky během prvního roku života. Kishnani PS et al J Pediatr 2004;144(5 Suppl):S35-S43. U starších dětí a dospělých je průběh onemocnění mnohem proměnlivější, zůstává však progresivní, což má za následek významnou morbiditu a často předčasnou mortalitu. Hagemans ML et al Brain 2005;128:671-7. Wokke J et al Muscle Nerve 2008;38:1236-45. Güngor D et al Orphanet J Rare Dis 2011; 6: 34. Pompeho nemoc je tedy stav s variabilní mírou progrese a nástupem symptomů. Všechny fenotypy Pompeho nemoci jsou způsobeny mutacemi v rámci stejného genu.
Průběh onemocnění u kojenců
Ve studiích znázorněných v níže uvedeném grafu byla diagnóza opožděna v průměru o téměř 3 měsíce, medián věku při úmrtí činil méně než 9 měsíců. van den Hout HMP et al Pediatr 2003;112(2):332-340. Kishnani PS et al J Pediatr 2006;148:671-676. To zdůrazňuje závažnost problému opožděné diagnózy u novorozenců s Pompeho nemocí vzhledem k rychlé progresi stavu v této populaci pacientů.
Věk nástupu symptomů, diagnózy a úmrtí u kojenců s Pompeho nemocí van den Hout HMP et al Pediatr 2003;112(2):332-340. Kishnani PS et al J Pediatr 2006;148:671-676.
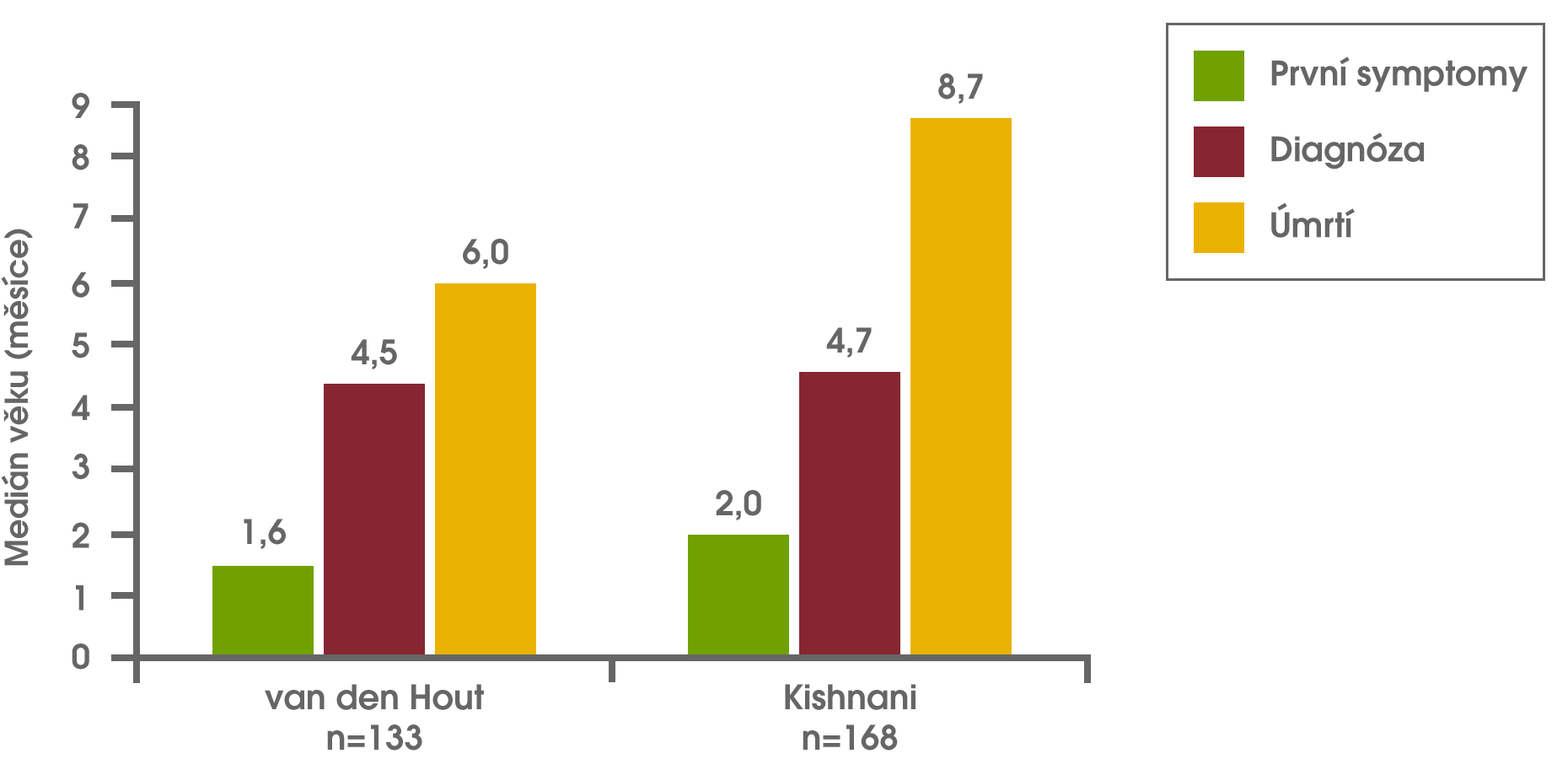
Převzato z: van den Hout et al, 2003 van den Hout HMP et al Pediatr 2003;112(2):332-340. a Kishnani et al, 2006. Kishnani PS et al J Pediatr 2006;148:671-676.
Průběh nemoci u dětí a dospělých
Hagemans et al hodnotili průběh Pompeho nemoci u 54 pacientů (průměrný věk 48,6 roků, věkové rozmezí 3,9 - 81,2 roků). Hagemans ML et al Brain 2005;128:671-7. Z toho u 40 pacientů byla předložena informace o době mezi první návštěvou lékaře kvůli potížím souvisejících s Pompeho nemocí a stanovením diagnózy tohoto stavu. U 28 % pacientů trvalo stanovení správné diagnózy 5 až 30 let, dalších 20 % čekalo na stanovení diagnózy 1 až 5 let. Hagemans ML et al Brain 2005;128:671-7. Tyto údaje slouží jako příklad opožděné diagnózy u dětí a dospělých, a vzhledem k neúprosné progresi Pompeho nemoci dokládají význam včasné diagnózy pro optimální management tohoto onemocnění.
Věkové rozložení specifických událostí při rozvoji Pompeho nemoci Hagemans ML et al Brain 2005;128:671-7.
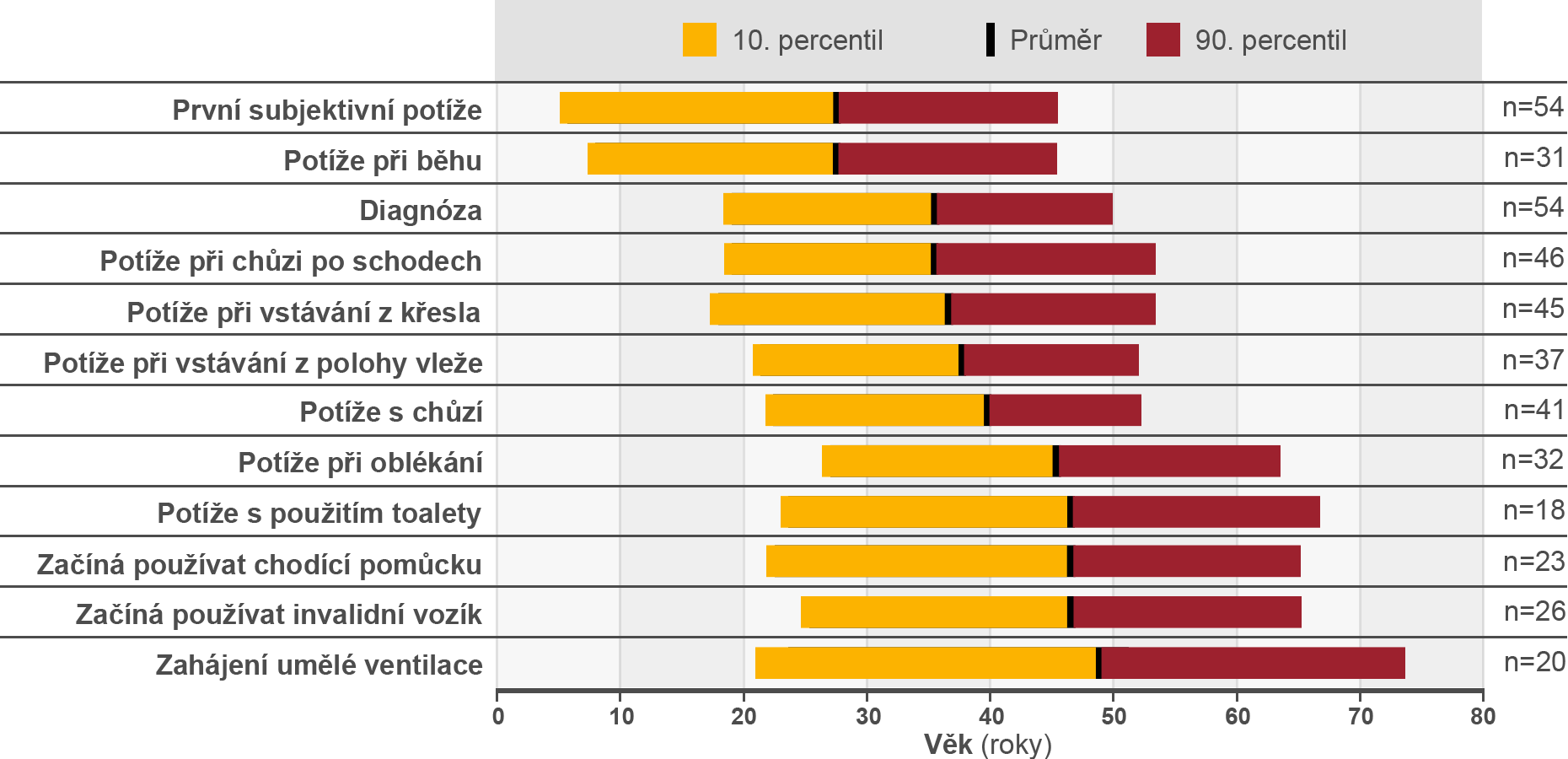
Převzato z: Hagemans et al, 2005. Hagemans ML et al Brain 2005;128:671-7.