

Diagnostické a konfirmační vyšetření
Pompeho nemoc je vzácné onemocnění s příznaky a projevy, které napodobují řadu jiných stavů. To znamená, že Pompeho nemoc má tendenci být přehlížena, minimálně zpočátku, dokud nejsou vyloučena jiná běžná onemocnění.
Výsledkem může být diagnostické opoždění. Včasná diagnóza je zvláště důležitá u kojenců, protože bez léčby obvykle dochází k úmrtí v průběhu prvního roku života. Retrospektivní analýza dětí v kojeneckém věku s Pompeho nemocí zjistila rozdíl 2,7 měsíce mezi mediánem věku při nástupu symptomů a mediánem věku při diagnóze. Kishnani PS et al J Pediatr 2006;148:671-676.
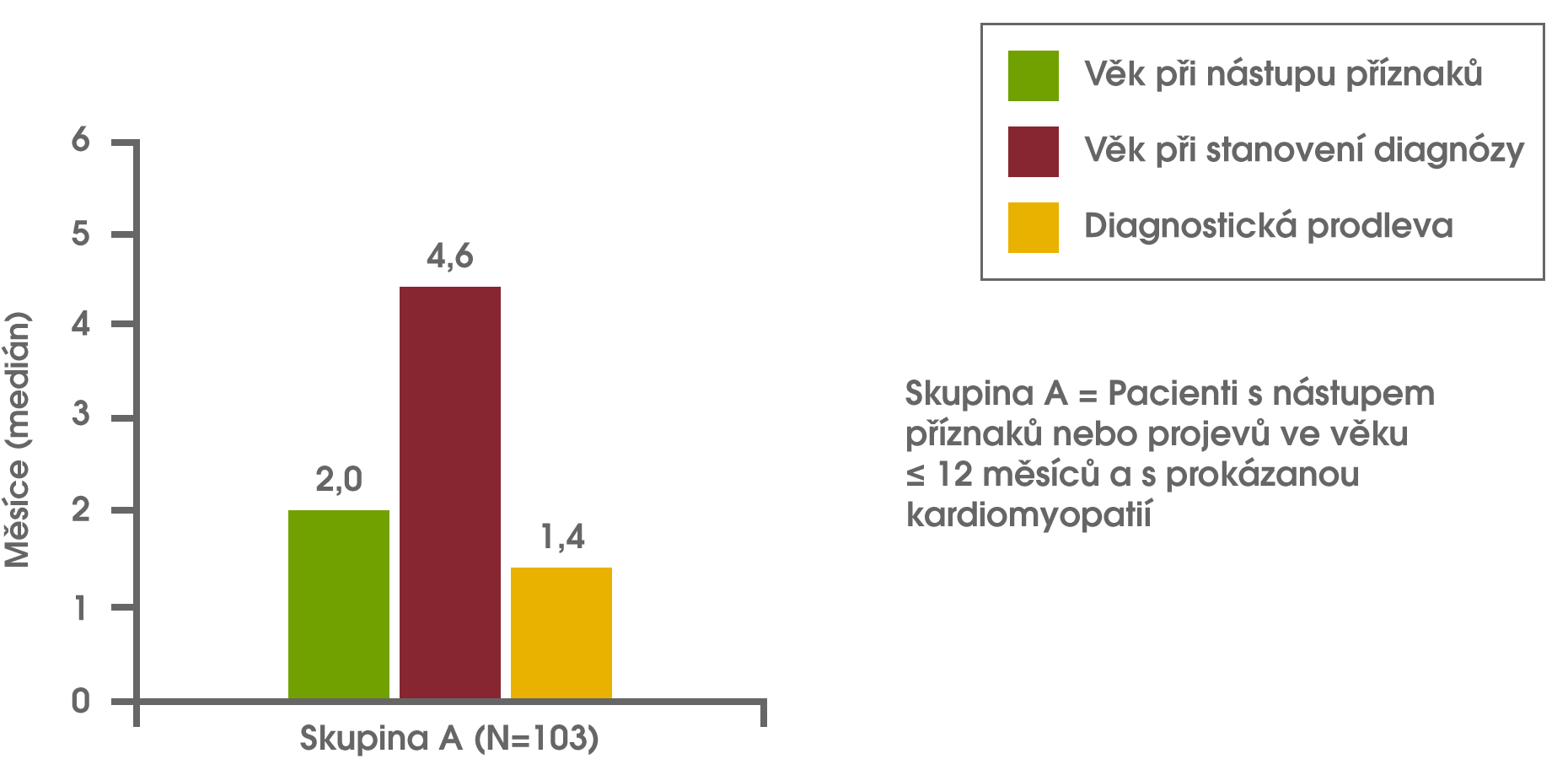
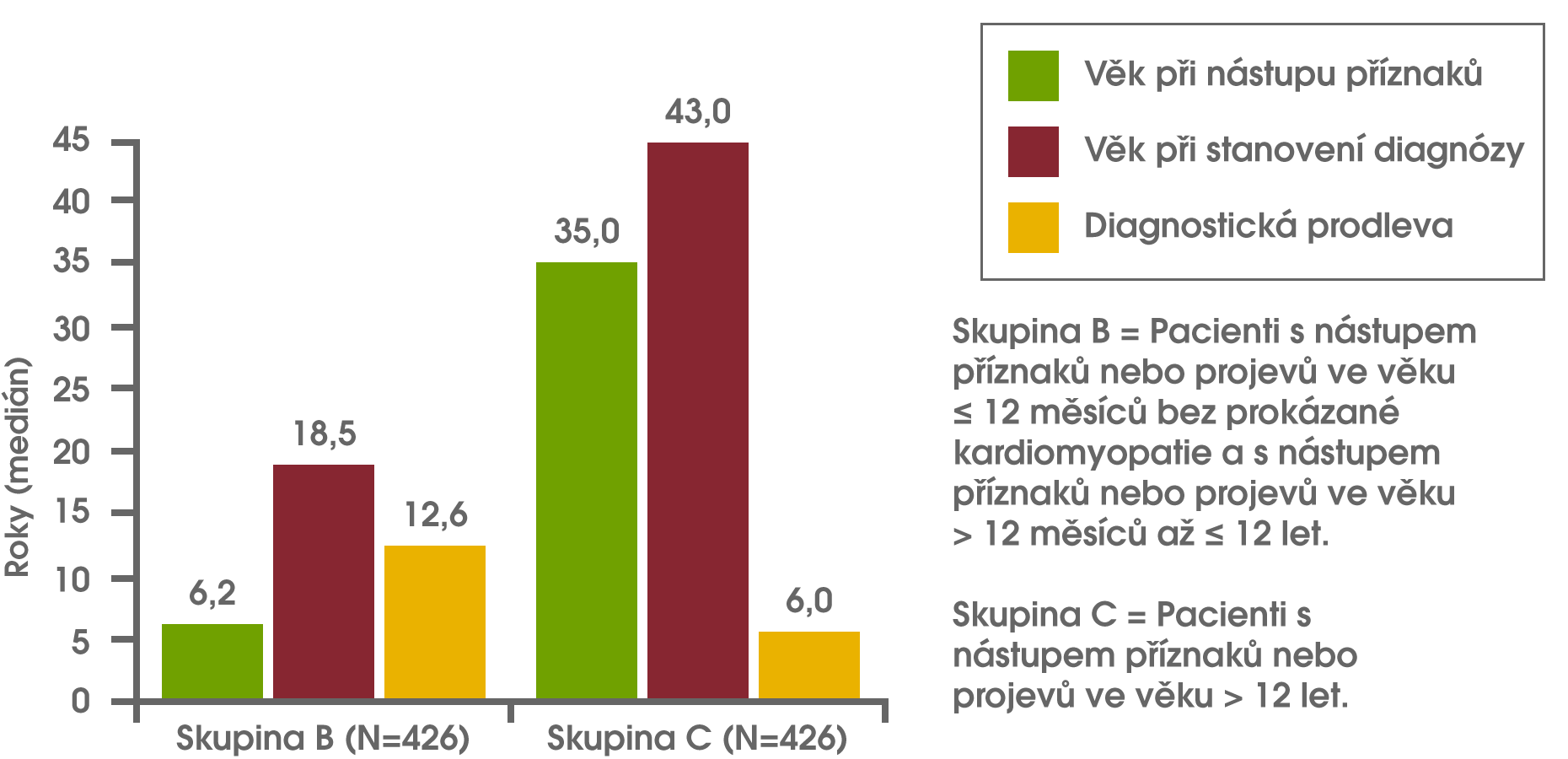
Analýza dat z registru Pompeho nemoci zjistila diagnostickou prodlevu u kojenců, dětí a u dospívajících/dospělých. U kojenců, u nichž došlo k nástupu symptomů během prvních 12 měsíců života a kteří byli léčeni kvůli kardiomyopatii (tj. klasická infantilní forma Pompeho nemoci), činil medián diagnostické prodlevy 1,4 měsíce. U pacientů s nástupem onemocnění po 12 letech věku činil medián diagnostické prodlevy 6 let. Nejdelší medián prodlevy v diagnóze činil 12,6 roku, a byl zaznamenán u pacientů s nástupem symptomů během prvních 12 měsíců života, u nichž však nebyla přítomna kardiomyopatie, nebo u pacientů s nástupem symptomů od 12 měsíců do 12 let věku. Kishnani PS et al Am J Med Genet A 2013;161A(10):2431-43.
Proto je zapotřebí zajistit časnější diagnózu napříč celým věkovým spektrem pacientů s Pompeho nemocí, a přispět tak k optimalizaci výsledků u pacientů.
Diagnostická prodleva u kojenců, dětí a dospívajících/dospělých s Pompeho nemocí Kishnani PS et al Am J Med Genet A 2013;161A(10):2431-43.
Definitivní diagnóza
Pompeho nemoc je potvrzena úplnou nepřítomností nebo výrazným snížením aktivity kyselé alfa-glukosidázy (GAA). Zhang H et al Genet Med 2006;8:302-306. Winchester B et al Mol Genet Metab 2008;93(3):275-281. Reziduální aktivita GAA u pacientů s Pompeho nemocí může činit méně než 1 % (obecně u dětí) až 30 % průměrné normální hladiny. van der Ploeg AT, Reuser AJ Lancet 2008;372(9646):1342-53. Pompeho nemoc může být rovněž potvrzena mutační analýzou, která potvrdí přítomnost dvou mutovaných alel (čtěte "Genetika a epidemiologie" ). van der Ploeg AT et al Eur. J. Neurol 2017; 24(6):768-e31.
Historicky bylo stanovení enzymatické aktivity GAA prováděno na kulturách kožních fibroblastů. Zhang H et al Genet Med 2006;8:302-306. Winchester B et al Mol Genet Metab 2008;93(3):275-281. Nicméně odběr vzorků je poměrně invazivní a získání výsledků trvá přibližně 6 týdnů. Tato dlouhá doba je zcela jednoznačně nežádoucí, a to zejména u dětí s rychle progredujícím onemocněním. Standardní praxí je dnes již použití krevních vzorků, včetně metody suché kapky krve. Odběr vzorků krve pro vyšetření na Pompeho nemoc je minimálně invazivní, přesné a zpravidla může poskytnout výsledky během několika dnů. Winkel LP et al J Neurol 2006;252:875-84. American Association of Neuromuscular & Electrodiagnostic Medicine Muscle Nerve 2009;40(1):149-160. V případě zjištění snížené enzymatické aktivity GAA je tento nález třeba potvrdit na druhém vzorku a/nebo sekvenováním genu pro GAA. American Association of Neuromuscular & Electrodiagnostic Medicine Muscle Nerve 2009;40(1):149-160.
Svalová biopsie je jednou z možností vyšetření aktivity GAA, která obecně není preferována. Důvodem této skutečnosti je invazivita a vysoké riziko falešně pozitivních výsledků v důsledku špatného zacházení se vzorky. Svalová biopsie může být užitečná pro histologické vyšetření, je však důležité si uvědomit, že obsah glykogenu se může u svalů výrazně lišit, a zdánlivě normální biopsie tedy nevylučuje Pompeho nemoc. Winchester B et al Mol Genet Metab 2008;93(3):275-281. Diagnóza Pompeho nemoci by proto měla být vždy potvrzena nepřítomností a/nebo sníženou aktivitou GAA nebo genetickou analýzou. van der Ploeg AT et al Eur. J. Neurol 2017; 24(6):768-e31.