

Klinický obraz
MPS I postihuje mnoho orgánových systémů a její fenotyp se mezi jednotlivými pacienty liší. Onemocnění vykazuje široké spektrum stupňů závažnosti, přičemž věk propuknutí příznaků se pohybuje od kojeneckého věku do dospívání. Onemocnění MPS I se z historického hlediska dělilo na tři subjektivně definované fenotypy, sahající od závažného fenotypu s rychlým postupem (syndrom Hurlerové) po mírnější fenotypy s pomalejším postupem (syndrom Hurlerové-Scheieův a Scheieův syndrom). Symptomatologie jednotlivých fenotypů se však značně překrývá a každý postižený jedinec vykazuje jiný klinický průběh a jedinečný (unikátní) soubor příznaků. Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123:19-29. Beck M et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. 2014;16(10):759-765.
Příznaky a projevy
Spektrum onemocnění MPS I


Informace v tabulce jsou čerpány z registru MPS.
Nejčastější projevy MPS I
Nejčastější projevy MPS I Michalík J, Zeman J et al. Mukopolysacharidóza. Olomouc: Společnost pro mukopolysacharidosu, 2010. ISBN 978-80-86417-11-0.

Víc informací o projevech MPS I (čtěte "Projevy nemoci" ).
Klinické projevy MPS I v číslech
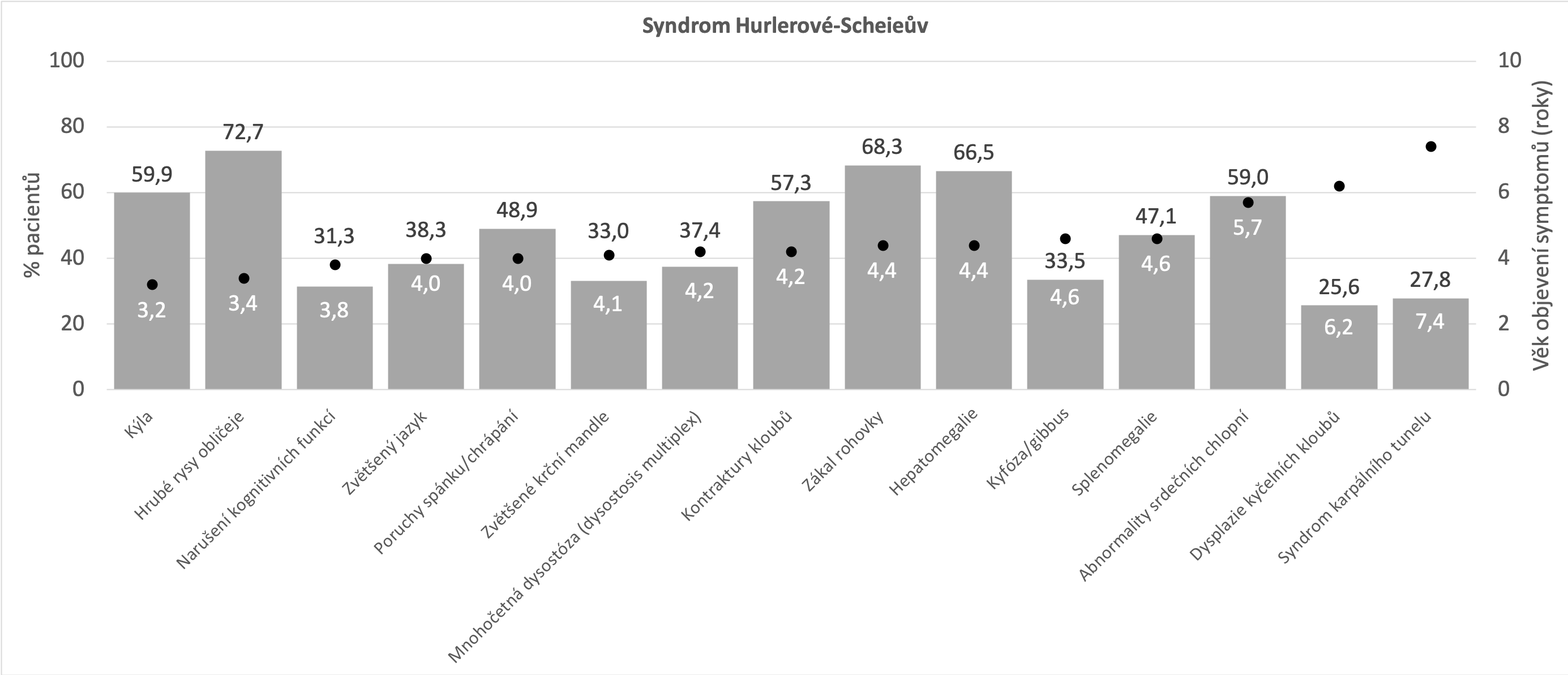
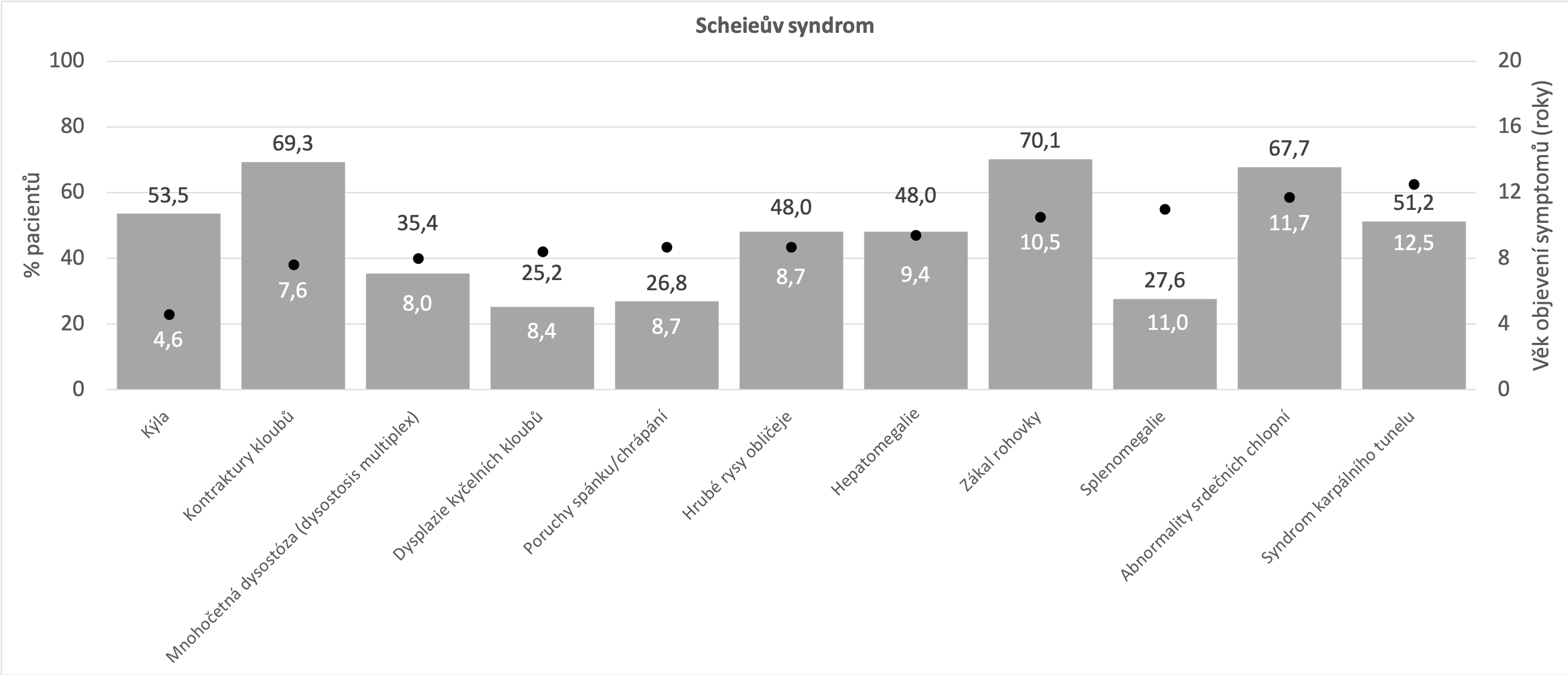
Níže uvedené grafy vycházejí z údajů, které byly shromážděny registrem MPS I o přirozeném průběhu onemocnění (Beck et al 2014). Tato analýza zahrnovala devět set padesát pět pacientů s následujícím rozložením fenotypů: syndrom Hurlerové - 601 (60,9%), syndrom Hurlerové-Scheieův - 227 (23,0%) a Scheieův syndrom - 127 (12,9%). Beck M et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. 2014;16(10):759-765.
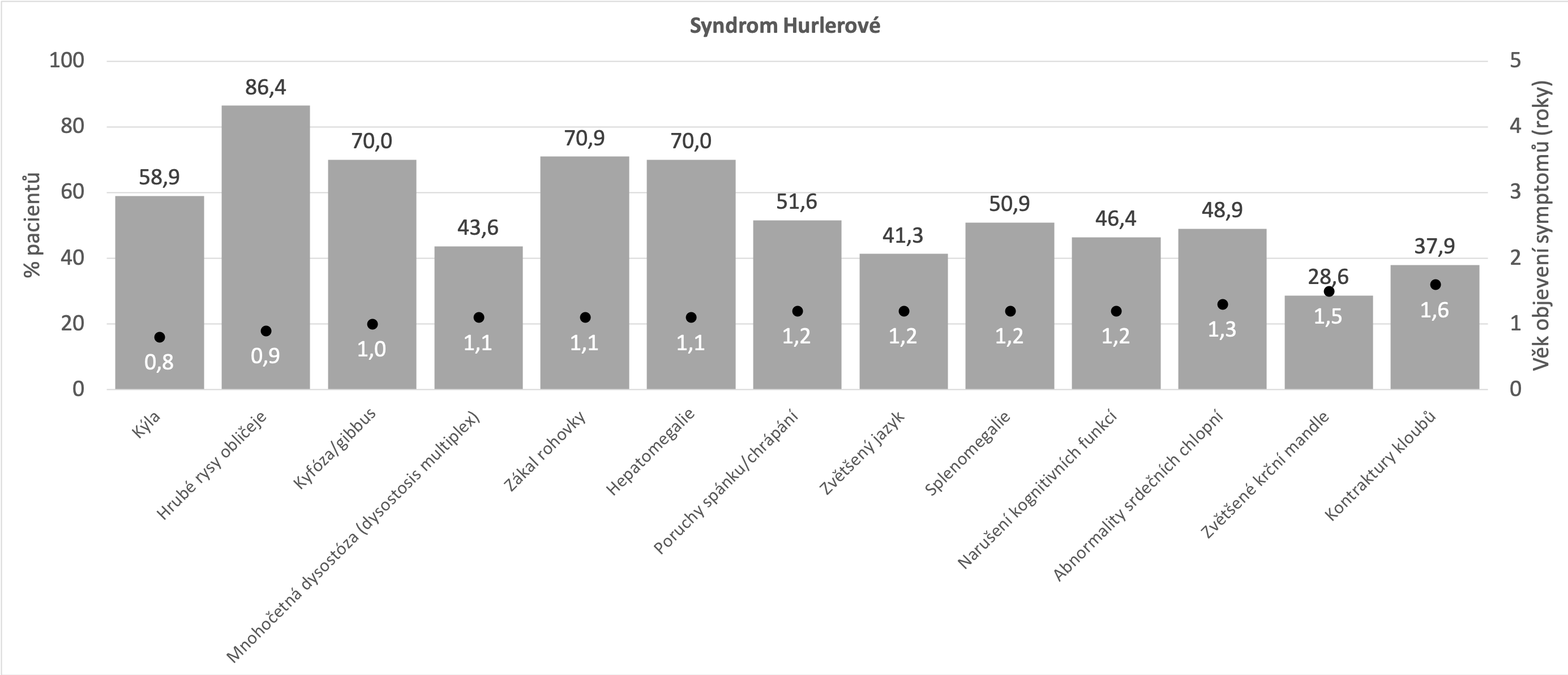

Těžká forma MPS I (syndrom Hurlerové)
Aleksandra | těžká forma MPS I (syndrom Hurlerové) | Polsko

Nejtěžší forma, tzv. Hurlerové syndrom, je multisystémové progredující onemocnění zahrnující typickou kraniofaciální dysmorfii, postižení skeletu charakteru dysostosis multiplex, zákal rohovky, psychomotorickou retardaci, četné kýly a hepatosplenomegalii. Děti s MPS I se rodí asymptomatické a již během kojeneckého období dochází k manifestaci prvních příznaků, které pozvolna progredují. Ješina P. Mukopolysacharidóza I v revmatologické ambulanci. Acta Medicinae 2016;7:3-6. Jejich stav se rychle zhoršuje, což vede k tomu, že většina neléčených pacientů během prvních deseti let života zemře. Beck M et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. 2014;16(10):759-765. U kojenců s MPS I jsou často popisovány makrocefalie, mírná hypotonie, hepatosplenomegalie, pupeční a tříselná kýla. Později v batolecím věku se prohlubuje kraniofaciální dysmorfie s prominujícím čelem, sedlovitým nosem, makroglosií, gingivální hyperplazií a zašpičatělými zuby s vrozenou mezerou mezi předními řezáky). Zpomaluje se růst, objevuje se disproporce těla s krátkým krkem, protruzí sterna a obvyklou hyperkyfózou v oblasti hrudní páteře. Ješina P. Mukopolysacharidóza I v revmatologické ambulanci. Acta Medicinae 2016;7:3-6.
Nejčastější projevy* Beck M et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. 2014;16(10):759-765.
- Opakovaná kýla (pupeční/tříselná)
- Deformita páteře - kyfóza/gibbus
- Hrubé rysy obličeje
- Zákal rohovky
- Hepatosplenomegalie
- Narušení kognitivních funkcí
- Poruchy spánku/chrápání
- Abnormality srdečních chlopní
- Nízký vzrůst
*ne všechny příznaky a projevy se musí vyskytovat u každého z pacientů
Mírnější forma MPS I (syndrom Hurlerové-Scheieův a Scheieův syndrom)
Ben │ mírnější forma MPS I (Scheieův syndrom) │ Velká Británie

Na druhé straně spektra fenotypů MPS I stojí Scheieův syndrom. Pacienti vykazují méně zjevné fyzické abnormality a mírné nebo žádné narušení kognitivních funkcí. Patří mezi ně drobné změny na skeletu a pojivové tkáni, charakterizované tzv. drápovitou rukou anebo syndromem karpálního tunelu, který není běžně popisován v dětském věku. Jeho přítomnost by měla vždy vést k podezření na MPS. Udává se, že přibližně polovina dětských pacientů se syndromem karpálního tunelu trpí některým z typů MPS I. Kardiologické projevy se omezují na postižení chlopní, nejčastěji aortální a mitrální, a často nejsou přítomny vůbec. Respirační obtíže jsou spojovány se spánkovou apnoí. Pacienti s tímto typem MPS I mají normální psychomotorický vývoj a zachován intelekt. Projevy postupují pomaleji než u těžce postižených pacientů. Příznaky, jako jsou různé typy kýly, kontraktury kloubů a/nebo respirační infekce, se obvykle objevují během prvních deseti let života, avšak mohou se podobat příznakům jiných častých onemocnění (například juvenilní a revmatoidní artritida Vijay S, Wraith JE. Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. ActaPaediatr 2005;94:872–877. - u pacientů, kteří vykazují kontraktury kloubů bez přítomnosti zánětu, by měl být proveden test na přítomnost MPS I Lehman TJA, Miller N, Norquist B et al. Diagnosis of the mucopolysaccharidoses. Rheumatology 2011;50:v41-v18. ), čímž dochází k léta trvajícímu oddálení stanovení diagnózy. Ješina P. Mukopolysacharidóza I v revmatologické ambulanci. Acta Medicinae 2016;7:3-6. Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001:3421-3452. Michalík J, Zeman J et al. Mukopolysacharidóza. Olomouc: Společnost pro mukopolysacharidosu, 2010. ISBN 978-80-86417-11-0.
Nejčastější projevy* Beck M et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. 2014;16(10):759-765.
- Opakovaná kýla (pupeční/tříselná)
- Kontraktury kloubů
- Syndrom karpálního tunelu
- Zákal rohovky
- Abnormality srdečních chlopní
- Poruchy spánku/chrápání
- Hepatomegalie
- Hrubé rysy obličeje
- Nízký vzrůst
*ne všechny příznaky a projevy se musí vyskytovat u každého z pacientů